FDA Enforcement Trends: Reflecting on 2019 and Looking Onward to 2020
Client Alert | 6 min read | 03.16.20
Warning Letters
This article is the third installment of a four-part series which leverages available FDA enforcement data from 2016 to present, with an emphasis on the pharmaceutical and medical device industries, to provide companies with insight on how to best comply with FDA regulations and avoid common pitfalls in 2020. The first installment in this series focused on trends in FDA inspections. The second installment focused on Form 483 Inspectional Observations. This article will address FDA Warning Letters which are formal notifications issued by senior FDA officials to notify a company of violations of regulatory significance. FDA considers this to be the Agency’s primary means to achieve prompt, voluntary compliance and ensure that management allocates appropriate resources to fully correct violations and prevent recurrence.
FDA Enforcement Tool Belt
As discussed previously, FDA has a variety of enforcement tools designed to encourage and compel compliance including Form 483s, warning letters, seizures, injunctions, criminal prosecution, and fines.
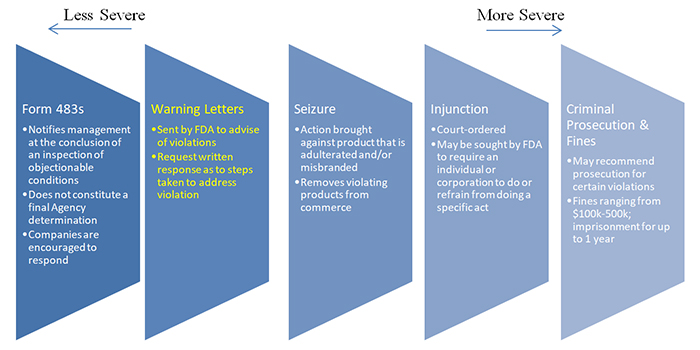
As illustrated in the graphic above, warning letters are considered an escalation from Form 483s and are often (but not always) issued after a company has failed to remedy violations noted in a Form 483 observation. They can be especially damaging to drug manufacturers as FDA typically will not approve new drug applications until it is satisfied that all issues at a facility have been fully addressed.
Manufacturers are expected to promptly rectify any regulatory violations described in a warning letter and provide a written response within 15 days. Note, however, that the response deadline may be expedited depending on the seriousness of the violations.
Ongoing or promised corrective actions, such as corrective actions a manufacturer may include in a 483 response, generally do not preclude the issuance of a warning letter. In determining whether to issue a warning letter after a corrective action has been undertaken or promised, the FDA will consider:
- The manufacturer’s compliance history (e.g., history of serious violations or failure to prevent the recurrence of violations);
- The nature of the violation (e.g., a violation that the manufacturer was aware of but failed to correct);
- The risk associated with the product and the impact on the violations on that risk;
- The overall adequacy of the proposed or ongoing corrective action and whether the corrective action addresses the specific violations, related violations, related products or facilities, and contains provisions to ensure effectiveness and prevent recurrence;
- Whether the manufacturer provided adequate documentation to allow the FDA to verify the proposed or ongoing corrective actions;
- Whether the timeframe for the corrective action is appropriate and/or feasible in light of the actual progress that has been made; and
- Whether the corrective action ensures sustained compliance.
Generally, FDA will not issue a warning letter if it concludes that the corrective actions are adequate and violations have been corrected.
Warning Letter Trends
Like FDA inspections, the number of warning letters issued to drug and medical device manufacturers decreased from Fiscal Years 2016 – 2019. There were 232 warning letters issued in 2016, 204 warning letters issued in 2017, 183 warning letters issued in 2018, and 162 warning letters issued in 2019.
The most common warning letter subject in FY 2019 for drugs and medical devices was for violations of current Good Manufacturing Practices (cGMP). The cGMP violations most commonly cited were:
- Adulterated and/or misbranded finished pharmaceuticals;
- Adulterated medical devices; and
- Adulterated Active Pharmaceutical Ingredients (API).
Although we are only a few months into 2020, the first warning letters issued to drugs and medical device companies may provide valuable insight into the Agency’s priorities this year. Based on available information, there have been 35 warning letter issued between January – February 2020.
Of these warning letters sixteen (16) have been issued to drug and medical device companies – four (4) warning letters issued to medical device manufacturers and twelve (12) issued to drug manufacturers. Nine (9) warning letters were issued to foreign drug or medical device companies, the majority of which followed the manufacturer’s placement on import alert. Nearly every warning letter indicated that the manufacturer’s response to the Form 483 was inadequate because it failed to address fully the scope of the observed problem and its potential impact to product on the market.
Examples of 2020 warning letter citations for drug manufactures include:
- Inadequate testing of finished pharmaceutical products (21 CFR 211.165(a) and (b)): A contract manufacturer of an over-the-counter drug product released drug products without conducting adequate testing, including identity and strength testing of the active ingredient, or appropriate testing for total aerobic microbial count and objectionable microorganisms. In response to the 483, the manufacturer stated that all batches of finished products with a particular active ingredient would be subjected to lab analysis prior to distribution. However, the FDA found the manufacturer’s response inadequate as the manufacturer failed to commit to performing assay testing and did not include information about the testing procedures, methods, timeline for implementation, or a detailed description of the tests it planned to conduct.
- Inadequate component testing (21 CFR 211.84(d)(1) and (2)): A drug manufacturer failed to adequately test incoming components, including the active ingredient, for their identity, strength, purity, etc. and, instead, relied on the supplier’s Certificates of Analysis (COA) without validating the supplier’s test results at appropriate intervals. FDA found the manufacturer’s 483 response to be inadequate as it failed to provide a comprehensive and retrospective assessment of all over-the-counter products which had been manufactured with components that had not been adequately tested to ensure adherence to quality attribute specifications.
- Failure to establish adequate written procedures for production and process control (21 CFR 211.100 (a) and (b)): Drug manufacturers failed to establish an adequate process validation program to evaluate the soundness of design and state of control of a process throughout the lifecycle of the drug product. FDA explained that one particular manufacturer’s procedure did not require an investigation and identification of root causes when products failed to meet quality requirements, and allowed for repeat testing and adjustments until the manufacturer achieved passing results. In response to the 483, the manufacturer stated that “the production process [could not] be validated” and “… small deviations are considered acceptable, as they do not influence the general usability of the product.” FDA noted that its response was inadequate because the manufacturer failed to commit to perform appropriate validation for each of its products and to fully remediate its systems for investigations of deviations.
2020 warning letter citations for medical device manufactures include:
- Failure to establish adequate design controls (21 CFR 820.30(a)): Medical device manufacturer was cited for failing to establish and maintain procedures to control the design of the device in order to ensure that design requirements are met. The warning letter explains that the manufacturer had distributed a medical device since September of 2018, prior to establishing a Design History File (DHF) that complies with FDA’s design control requirements.
- Failure to establish and maintain procedures for validating the device design that includes risk analysis, where appropriate (21 CFR 820.30(g)): FDA noted that the manufacturer’s 483 response was inadequate, in part, because while the manufacturer provide a CAPA to address the deficiency and represented that it was in the process of implementing the corrective actions, the manufacturer failed to provide confirmation that the proposed corrective actions were appropriate and effective.
- Failure to establish and maintain procedures to control product that does not conform to specified requirements, including a determination of the need for an investigation and documentation of that investigation (21 CFR 820.90): FDA has found 483 responses to be inadequate, in part, when the company has failed to properly review the history of nonconformity of the devices it manufactures.
Your Company Has Received a Warning Letter – Now What?
In many respects, responding to a warning letter is similar to a Form 483 response. It is important to respond promptly (within 15 business days), thoroughly, with consideration of the broad impact of FDA’s stated concerns, and carefully.
The warning letter, however, is indication that FDA is not satisfied with the original response, most commonly because the Agency does not think it went far enough. It is important, therefore, to consider carefully where the Form 483 response may have fallen short, recognizing that FDA won’t necessarily be explicit. Was adequate consideration given to all affected product? Is there product on the market that should be recalled? Is it necessary to bring in independent auditors to evaluate a corrective action? A thorough and broad assessment is essential.
Conclusion
FDA’s warning letters have been on the decline, and while this may continue through 2020, we anticipate foreign manufacturing facilities will continue to be a focus of FDA enforcement activities. Therefore, it is critical for manufacturers, especially foreign ones, to take every precaution to limit the risk of receiving a warning letter. Likewise, U.S. distributors should exercise appropriate oversight over foreign manufacturers to not only avoid supply disruption, but to reduce their own risk of FDA enforcement actions.
The next and final article will address the most serious FDA enforcement actions – seizures, injunctions, criminal prosecution, and fines.
Contacts

Insights

Client Alert | 7 min read | 06.24.26
On June 17, 2026, the U.S. Department of Justice’s (DOJ( National Security Division (NSD) announced that it had issued a declination for Robert Bosch GmbH (Bosch) relating to potential violations of the Export Control Reform Act, 50 U.S.C. § 4819 (ECRA). Specifically, the DOJ declined to criminally prosecute Bosch’s violations of the Export Administration Regulations’ (EAR) Foreign Direct Product Rule (FDPR), which apparently resulted from two Bosch subsidiaries’ export of products and software manufactured with equipment that was the direct product of U.S. software or technology to Huawei Technologies Co., Ltd. and its “Entity List” affiliates, including Huawei Tech. Investment Co., Ltd., Hong Kong (collectively, Huawei). The same day, the U.S. Department of Commerce Bureau of Industry and Security (BIS) announced a parallel civil administrative settlement with Bosch.
Client Alert | 4 min read | 06.23.26
EPA Hands Over AI Data Center Regulation to States and Communities to Develop Best Practices
Client Alert | 3 min read | 06.22.26
Client Alert | 4 min read | 06.17.26
From Checkout To Opt-Out: The EU Withdrawal Button Is Here – What E-Commerce Businesses Need To Know