FDA Enforcement Trends: Reflecting on 2019 and Looking Onward to 2020
Client Alert | 6 min read | 02.12.20
Form 483 Observations
This article is the second installment of a four-part series which leverages available FDA enforcement data from 2016 to present, with an emphasis on the pharmaceutical and medical device industries, to provide companies with insight on how to best comply with FDA regulations and avoid common pitfalls in 2020. The first installment in this series focused on trends in FDA inspections. This article will focus on the most common FDA enforcement action that may follow from an inspection, Form 483 Inspectional Observations – most commonly referred to in the industry as simply “483s.”
FDA Enforcement Tool Belt
As discussed in the first installment of this series, the FDA has a variety of enforcement tools designed to encourage and compel compliance with regulations, including Form 483s, warning letters, seizures, injunctions, criminal prosecution and fines.
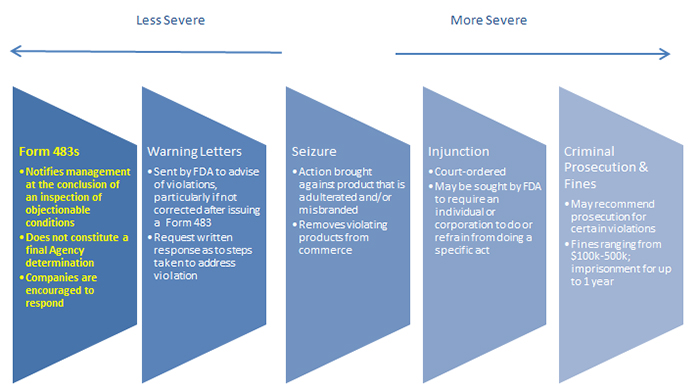
As shown in the graphic above, Form 483s are the least severe FDA enforcement mechanism – and they are also the most common. However, there is no such thing as a “minor” Form 483 observation. By definition, FDA considers any observation it chooses to document in a Form 483 to be a serious violation of FDA regulations, one that could result in escalated regulatory action if not promptly addressed. Thus, companies should take all Form 483 Observations seriously.
Form 483s are issued and discussed at the close of an inspection and list conditions observed by the FDA investigator(s) that, in their judgment, constitute significant violations of the Food, Drug, and Cosmetic Act (FDCA). While 483s note only significant violations of the FDCA, they are not final agency determinations. In addition, companies are not required to respond – but they absolutely should. The response should be in writing and include the company’s corrective action plan. Importantly, the corrective action plan should be promptly implemented.
Form 483s can also have negative litigation implications for all FDA-regulated companies. Although Form 483s are not a final agency determination, Plaintiff’s lawyers may nonetheless seek to use the findings against a company in products liability lawsuits to support their claims that a company was not manufacturing products in compliance with FDA regulations.
Looking Back: Form 483 Observation Data
In our first installment, we noted that the number of FDA inspections is on the decline. While inspection numbers are dropping, the number of Form 483 Observations has remained relatively consistent, according to available FDA data.1
Table 1. Total Number of Form 483 Observations for Fiscal Years 2016-2019
FY 2016 |
FY 2017 |
FY 2018 |
FY 2019 |
|
4,528 |
5,045 (+11.4% from 2016) |
4,910 (-2.7% from) 2017) |
4,770 (-2.9% from 2018) |
The percentage of Form 483 Observations attributable to drug and medical device manufacturing also remained consistent from 2016 to 2019 – with drug-related observations ranging between 14-16% of all observations, and medical device-related observations ranging between 17-21% of all observations. FDA has also focused on the same types of FDCA violations by drug and medical device manufacturers year after year – which provides insight into how companies can best prepare for upcoming inspections.
The following three observations topped the list of drug-related observations for FY 2016-2019:
- Quality control procedures not in writing, fully followed [21 CFR § 211.22 (d): “The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed.”]
It is not enough to simply make a quality product. When FDA inspectors visit your facility, it’s imperative that you are able to demonstrate compliance with CGMPs through readily accessible, clearly written, and consistently maintained and modified Standard Operating Procedures (SOPs). - Scientifically sound laboratory controls not established [21 CFR § 211.60(b): “Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling, and drug products conform to appropriate standards of identity, strength, quality, and purity.”]
FDA is a science-based agency and, thus, will expect that your facility is relying on and applying good scientific principles and practices. - Investigations of discrepancies, failure to adequately review [21 CFR § 211.192: “All drug product production and control records, including those for packaging and labeling, shall be reviewed and approved by the quality control unit to determine compliance with all established, approved written procedures before a batch is released or distributed. Any discrepancy (including a percentage of theoretical yield exceeding the maximum or minimum percentages established in master production and control records) or the failure of a batch or any of its components to meet any of its specifications shall be investigated, whether or not the batch has already been distributed.]
Laboratories should have a scientifically validated investigation strategy. For example, the laboratory should not simply re-test or test products into compliance. All out-of-spec (OOS) test results should be investigated to identify the root cause – whether or not the subject product or material is actually distributed.
The following two observations have topped the list of medical device-related observations for FY 2016-2019:
- Lack of or inadequate procedures (CAPAs): [21 CFR § 820.100(a): “Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for: (1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product . . . (2) Investigating the cause of nonconformities relating to product, processes, and the quality system; (3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems; (4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems; (6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and (7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.”]
During an FDA inspection of a medical-device facility, the CAPA (corrective action and preventive action) processes will be scrutinized. Companies should take proactive steps to ensure that its approach to initiating CAPAs is effective and followed. - Lack of or inadequate complaint procedures [21 CFR § 820.198(a): “Each manufacturer shall maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit. Such procedures shall ensure that: (1) All complaints are processed in a uniform and timely manner; (2) Oral complaints are documented upon receipt; and (3) Complaints are evaluated to determine whether the complaint represents an event which is required to be reported to FDA under part 803 of this chapter, Medical Device Reporting.”]
Medical device facilities should promptly investigate complaints and document the entire complaint handling process. The complaint handling process is imperative as it provides the data necessary for a company to determine if its medical device(s) present potential risks to patients. During an inspection, FDA investigators will take a close look at the facility’s complaint procedures to ensure that it maintains records of complaints received and has procedures in place for receiving, reviewing and evaluating complaints.
Looking Ahead to 2020: How to Respond in the Event of a Form 483
Given the consistency of available data, it is likely that the same Form 483 Observations will remain a high priority for the agency in 2020 and beyond. While Form 483s are the least severe FDA enforcement mechanism, it is crucial that companies appropriately respond to any Form 483 Observations and remedy all practices found to violate the FDCA. Repeat violations risk more damaging action by the agency.
Below are a few tips for responding to 483s to reduce the likelihood of receiving a warning letter in 2020:
- Respond promptly. FDA expects companies to respond to Form 483 Observations within 15 business days.
- Respond thoroughly. Keep in mind that the audience for your responses is not limited to FDA investigators, but includes compliance officers, program officers and perhaps even lawyers and judges who have never been to your facility. Your response needs to provide a complete picture of the issue raised in the observation, putting it in proper context.
- Consider the broad impact. FDA investigators may inspect records pertaining to a single lot. If they find a problem, however, they will not assume it was an isolated case. Any investigation and response must consider the impact of the problem on other lots and, most importantly, product that is out on the market.
- Respond carefully. Responses to Form 483 Observations are representations to the government. They must be accurate and set reasonable expectations. As noted, responses may also become evidence in product liability actions. The company must be able to defend the statements it makes.
Conclusion
FDA has consistently returned to the same Form 483 Observations for drugs and medical device manufacturers over the past four years – and this trend is likely to continue, giving companies insight into how to be best prepared for their next inspection.
In the event your company receives a Form 483, it is crucial to respond appropriately, as repeat violations can lead to more severe enforcement actions by FDA – including the issuance of a Warning Letter or worse. Warning Letters will be the subject of the third installment in this four-part series.
1 U.S. Food & Drug Administration, Inspection Observations, https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations (last visited Jan. 1, 2020). Notably, this data reflects the total number of Form 483 observations issued by FDA, rather than the number of Form 483s, as multiple observations may be made in the same Form 483.
Contacts

Insights

Client Alert | 3 min read | 08.03.26
On July 20, 2026, New Jersey Governor Mikie Sherrill signed the Forbidding the Algorithmic Inflation of Rent (FAIR) Act into law, making New Jersey the fourth state to regulate algorithmic rent-setting practices. Three days later, on July 23, 2026, Governor Sherrill signed the Fair Price Protection Act, which targets “surveillance pricing”—the practice of using or collecting personal data about a user and using an algorithm or artificial intelligence to charge different consumers different prices for the same products. Together, these laws represent a significant expansion of New Jersey's consumer protection framework in the algorithmic pricing context.
Client Alert | 2 min read | 08.03.26
New York Becomes First State to Restrict Addictive Social Media Features for Minors
Client Alert | 4 min read | 08.03.26
Client Alert | 6 min read | 08.03.26
The Pipe, Not the Posts: How Section 230’s Protections Extend to Generative AI Platforms