Recent Happenings in Advertising & Product Risk Management – December 2017
Client Alert | 39 min read | 12.20.17
In this issue:
- Feature Article: FDA Issues First-of-Its-Kind Guidance for Manufacturers of 3D-Printed Medical Devices
- Article: California Updates Automatic Renewal Law for Subscription-Based Businesses
- Litigation Roundup
- Filed
- Several Major Retailers Implicated in Flushable Wipes Class Action Lawsuit
- “Groundbreaking” Allergan Lanham Act Suits Against Drug Compounders Survives Motion to Dismiss
- Settled
- Utz Settles “All-Natural” Class Action for $1.25M
- Data Breaches Cost Cottage Health $2M in Recent Settlement
- The Children’s Place Settles Deceptive Discount Pricing Class Action for at Least $6.8 Million
- Dismissed/Stayed
- Federal Judge Dismisses Dannon “Natural” Yogurt Labeling Suit
- FTC and NY AG’s False Advertising Claims Against Prevagen Dismissed
- Filed
- Regulatory Roundup
- CPSC
- FDA
- FTC
- NAD
- Crowell & Moring’s Retail & Consumer Products Law Observer
- ROSCA Enforcement Ahead: FTC Settles with AdoreMe for $1.38 Million
- “Do Not Resuscitate”: Lessons for Advertisers
- Soda Stays Safe in San Francisco
- Court Dismisses FTC’s Unfairness Claims Against D-Link
- Quincy Biosciences: What the Decision Means for Advertising of Health Claims, and What It Means to the FTC
- It’s Only A Game. Or Is It? Advergaming In The Digital Age
- Report on the Autonomous Vehicle Safety Regulation World Congress 2017
- Subverting Democracy, Advertising, and the Economy Through Bots
- Crowell & Moring Speaks
This news bulletin is provided by the Advertising & Product Risk Management Group of Crowell & Moring. If you have questions or need assistance, please contact Chris Cole, Cheryl Falvey, or any member of the APRM Group.
FEATURED ARTICLE
FDA Issues First-of-Its-Kind Guidance for Manufacturers of 3D-Printed Medical Devices
3D printing, commonly referred to as “additive manufacturing,” is an emerging area of technology poised to revolutionize the way commercial and consumer products are manufactured. This technology is relatively nascent, and U.S. regulatory bodies have not acted quickly to enact or modify regulations to specifically address 3D-printing technologies. Earlier this month, the U.S. Food and Drug Administration (FDA) became the first agency to speak comprehensively on its present thinking and expectations for regulation of 3D-printed products.
On December 4, 2017, the FDA issued first-of-its-kind guidance to manufacturers of 3D-printed products—in this case, medical devices.1 The FDA’s 28-page guidance offers manufacturers of 3D-printed medical devices a path to regulatory compliance. While this is likely only the beginning of FDA action regarding 3D-printing technologies, this guidance could have implications for other agencies considering regulations, and courts that may perceive this non-binding guidance to constitute the industry’s standard of care.
FDA’s Guidance
FDA’s technical guidance document is entitled “Technical Considerations for Additive Manufactured Medical Devices.”2 The FDA describes the guidance as being “broadly organized into two topic areas: Design and Manufacturing Considerations . . . and Device Testing Considerations.” Thus it speaks both to quality requirements, and is intended to clarify the pathway additive manufacturers must follow in seeking FDA approval to sell 3D-printed medical devices (but does not apply to biological, cellular or tissue-based products). The guidance is neither binding nor likely to be static. It outlines the Agency’s current thinking on approaches to additive manufacturing, including on such topics as general and patient-matched device design, product testing, and current good manufacturing processes.
In a statement announcing the final guidance,3 FDA Commissioner Scott Gottlieb characterized the document as both a “comprehensive technical framework” and “leap-frog guidance” offering the Agency’s “initial thoughts” on medical devices created by additive manufacturing. According to Commissioner Gottlieb, the FDA’s “recommendations are likely to evolve as the technology develops in unexpected ways.” “In order to ensure the safety and effectiveness of [3D printed medical devices], [the FDA is] working to establish a regulatory framework for how [it] plan[s] to apply existing laws and regulations that govern device manufacturing to non-traditional manufacturers like medical facilities and academic institutions that create 3D-printed personalized devices for specific patients they are treating.” Although the Commissioner did not articulate the FDA’s precise agenda when it comes to 3D-printed devices, he noted that FDA engineers in the Center for Devices and Radiological Health (CRDH) have been conducting research in their own 3D printing facility to determine the effect that changes in the production and design processes can have on a device’s safety and functionality. According to the Commissioner, “[t]his research . . . helps inform us as regulators to help us understand the policy framework needed to ensure the quality and safety of 3D printed products.”
While the guidance is not technically binding, it is final. This means it expresses current Agency expectations when evaluating quality practices and premarket submissions for 3D-printed medical devices that manufacturers should follow unless they can demonstrate that an alternative approach satisfies the requirements of the applicable statutes and regulations. Highlights of the guidance document include:
- Over-Arching Principle: FDA explains that “[i]t is anticipated that [additive manufactured] devices will generally follow the same regulatory requirements as the classification and/or regulation to which a non-[additive manufactured] device of the same type is subject.”
- Quality System: Manufacturers of medical devices made in whole, or in part, by additive manufacturing should follow Quality System (QS) requirements to ensure that the devices can perform as intended. Accordingly, manufacturers should identify risks and document how they intend to mitigate those risks throughout the manufacturing process, using “all reasonably obtained knowledge about [the 3D printing] machine’s capabilities to ensure the manufacturing process outputs meet defined requirements. Quantitative knowledge of the machine’s capabilities and limitations can be gained through test builds, worst-case builds, or process validation.”
The Agency further recommends using a production flow diagram that identifies the steps involved in the manufacturing of the device to help ensure that all elements of product quality are addressed. An example of an additive manufacturing production flow chart that appears in FDA’s guidance document is reproduced below.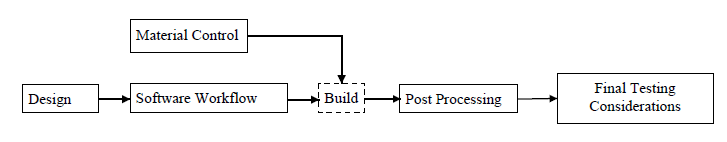
- 3D Printing Machines: To ensure that the product can be reliably built, FDA recommends that manufacturers compare and document the desired design of the device to the “manufacturing tolerances of the individual machine.” FDA also notes that the quality of medical products may vary when built using different additive manufacturing machines. “Therefore, knowledge of how the variability of each input parameter and processing step affects the final finished device or component is critical to ensuring part quality.”
- Material Controls: The Agency notes that materials may undergo significant chemical and/or physical changes in the manufacturing process, which could affect the integrity of the medical product. As such, manufacturers should implement appropriate material controls “[t]o ensure consistency of the incoming raw material and the final product.” In addition, manufacturers should understand and document the effects that material specification changes and material reuse have on the build process and final device.
- Post-Processing: FDA acknowledges that manufacturing steps conducted after the 3D printing process may impact the performance of the final device. It therefore recommends that additive manufacturers identify any potential detrimental effects and establish procedures to maintain device performance.
- Revalidation: FDA advises that “[c]hanges to the device, manufacturing process, or process deviations should be identified and analyzed for the potential risks they introduce.” Such changes or deviations may require the additive manufacturer to revalidate the manufacturing process. Examples of revalidation triggers include: software changes (e.g., changing or updating build preparation software); material changes (e.g., supplier, incoming material specification, reused powder, new formulation) or material handling; changes to the spacing or orientation of devices or components in the build volume; changes to the software workflow; and physically moving the printing machine.
The FDA emphasized that “[n]ot all considerations described will be applicable to every device, given the variety of [additive manufacturing] technologies, materials, and devices made with additive manufacturing.”
Will Other Agencies Follow Suit?
Although each federal agency acts independently, by issuing this guidance, FDA may have set a standard that influences other regulators. Historically, it is not uncommon for one agency’s thought leadership about a new technology or scientific advancement to influence other agencies’ thinking and action on those issues. For instance:
- In developing a summary of cybersecurity best practices in the automotive industry, the National Highway Traffic Safety Administration (NHTSA) looked to the National Institute of Standards and Technology (NIST) cybersecurity standards as a “baseline” for federal cybersecurity best practices and industry-specific security guidelines.4
- The Consumer Product Safety Commission (CPSC) has also followed the Environmental Protection Agency’s (EPA) lead on chemical risk assessments. For instance, in issuing its final rule prohibiting children’s toys and child care articles from containing specific phthalates, 16 C.F.R. part 1307, the CPSC justified the propriety of conducting a cumulative risk assessment in part by pointing to EPA’s routine use of cumulative risk assessments to assess certain chemicals.5
Implications in Medical Device Litigation?
FDA’s non-binding guidance may further influence judicial actions involving additive manufacturing, to the extent that courts treat the FDA’s guidance as evidence of the industry’s standard of care. It also would not be without precedent for courts to do so. In the food litigation context, for instance, courts have found that the FDA’s method of testing compliance also applies to plaintiffs in a private action, even though FDA’s regulations do not require private litigants to follow the same methodology. In fact, in February 2016, an MDL court dismissed claims asserted in 11 putative class action lawsuits alleging that the sugar content in Whole Foods’ store-brand yogurt was false and misleading because their evidence of a misstatement of the yogurt’s sugar content did not comply with FDA testing methodology.6
* * *
Even though the FDA’s guidance is “final,” stakeholders can still become engaged with the FDA as it assesses how to regulate 3D-printed medical devices. FDA’s good guidance practices require that the Agency continue to accept comments at any time, and a docket has been set up to receive comments on this guidance.
For more information, contact: Rebecca Chaney, Robbie Rogart, and Chalana Williams
ARTICLE
California Updates Automatic Renewal Law for Subscription-Based Businesses
With the boom in subscription-based businesses, California recently updated its Automatic Renewal Law (ARL), Cal. Bus. & Prof. Code § 17600 et seq. These new requirements will require subscription-based service providers to quickly adapt their current practices to avoid increased litigation or civil penalties under California’s newly amended ARL. The ARL was originally enacted in 2010 and was met with a flurry of litigation in California, including several class actions.
The updated law, effective on July 1, 2018, applies to businesses which offer an automatic renewal or continuous service, including free gift or trial offers. Automatic renewal offers must include “a clear and conspicuous explanation of the price that will be charged after the trial ends or the manner in which the subscription or purchasing agreement pricing will change upon conclusion of the trial.” Senate Bill No. 313 (Sept. 28, 2017).
When a service is offered at a promotional or discounted price for a limited period of time, the advertiser must first gain consent before charging a consumer’s credit or debit card. If the offer also includes a free gift or trial, the advertiser is required to disclose how the consumer can cancel the service prior to getting charged or before the expiration of the promotional rate. If the offer is made online, the advertiser must allow the customer to cancel online.
Subscription-based service providers should consider implementing the following best practices in advance of July 1, 2018:
- Use “clear and conspicuous” language, especially when communicating cancellation policies to consumers.
- Inform consumers of cancellation policies and any instructions for cancellation (again in “clear and conspicuous” language) prior to consumers purchasing any goods or services.
- Ensure that cancellations are relatively easy for the average consumer to accomplish.
- Consult with an experienced attorney on whether to include arbitration provisions (or other liability limiting language, such as a class-action waiver) in literature provided to consumers.
- Assure there are mechanisms in place to receive consumers’ affirmative consent prior to charging consumers.
- Consider including “exclusively online” cancellation features where applicable.
Subscription-based service providers have ample time, before July 1, 2018, to adapt to California’s new amendments; such providers should quickly innovate and implement new business practices that conform to California’s new amendments to its ARL.
For more information, contact: Rukiya Mohamed
LITIGATION ROUNDUP
The litigation arena for the consumer products industry is as active as ever. Each newsletter we bring you a summary of the most important litigation developments from the past two months, from complaint filings and key court decisions to trial results and settlements. For more information about these and other developments, please visit our Food & Beverage Industry Tracking Report.
Filed
Several Major Retailers Implicated in Flushable Wipes Class Action Lawsuit
On December 5, 2017, in a New York federal court, a new class action lawsuit alleged that retailers including Costco, Target, and Wal-Mart manufacture and sell wipes that are incorrectly marketed as safe to flush, causing considerable long-term damage to sewage systems. The party representing the class—a New York-based homeowners association that operates a sewage treatment plant claims that the operators of the facilities bear the cost of maintenance and repairs of the sewage systems caused by the wipes. They take issue with the fact that the wipes are labeled as “safe to flush, safe for plumbing, safe for sewer systems and/or biodegradable,” while they allegedly are not. The homeowners association seeks to represent a class of New York-based operators of sewage treatment plants and a separate class of all U.S. operators of sewage treatment plants that have been affected by the wipes since Dec. 4, 2011. They allege strict product liability for defective design, failure to warn, nuisance, trespass, breach of express and implied warranties, negligence, and negligent misrepresentation.
“Groundbreaking” Allergan Lanham Act Suits Against Drug Compounders Survives Motion to Dismiss
Two lawsuits filed in California federal court by drug company Allergan PLC in early September have been recently described as “seminal” and “groundbreaking,” in that they set the stage for testing to what extent drug compounders can mass-produce virtual copies of brand-name prescription drugs. They are among the first lawsuits where a large pharmaceutical company has used the Lanham Act to allege that compounders are falsely claiming to be in compliance with FDA regulations (rather than relying on government regulators to police activity). These suits coincide with litigation against the FDA by Par Pharmaceutical accusing the FDA of letting compounders sell large quantities of copycat drugs and skirting the FDA approval process. Recently, on December 4, 2017, Judge David O. Carter refused to dismiss the lawsuit.
Settled
Utz Settles “All-Natural” Class Action for $1.25M
On December 7, 2017, Utz Quality Foods settled a proposed class action launched back in 2014 for $1.25 million that asserted that several of their snack foods are deceptively labeled as “all natural.” Some of the products at issue include their brand of regular potato chips, honey wheat pretzel sticks, and restaurant-style tortillas. Plaintiffs alleged that these products, and others, contain unnatural genetically-modified, synthetic, and artificial ingredients, and thus, they had breached express warranty, engaged in deceptive acts or practices and misleading advertising, and had been unjustly enriched. Although Utz has agreed upon the payment, they “den[y] and continue[] to deny all liability with respect to any and all of the claims alleged.”
Data Breaches Cost Cottage Health $2M in Recent Settlement
In another recent data breach lawsuit, Cottage Health agreed to pay $2 million to settle claims that it failed to implement basic safeguards for patient data that led to two separate data breaches leaking medical information. The first breach released the private information of more than 50,000 patients from 2011–2012. The second breach in 2015 leaked more than 4,500 patients’ medical records. Attorney General Xavier Becerra announced the settlement on November 22. Cottage Health has agreed to implement upgrades including new system monitoring, firewalls, network intrusion detection, and access management protocols to protect patient data. It will also have to hire a data privacy security officer to ensure compliance and assist in completing annual privacy risk assessments.
The Children’s Place Settles Deceptive Discount Pricing Class Action for at Least $6.8 Million
On November 27, 2017, The Children’s Place agreed to settle a class action lawsuit launched in California federal court by plaintiffs claiming that the retailer offered “phantom markdowns.” They alleged that The Children’s Place inflated printed “original” prices in order to offer “sale” prices that seemed far greater than they were because the retailer rarely, if ever, offered the merchandise at the purported “original” prices. This class action has been one of several recently issued against retailers such as Gap, Amazon, Burberry, and Burlington Coat Factory, centering on deceptive discount pricing. The settlement also follows JC Penny’s $50 million settlement in 2016 and Michael Kors’ $4.9 million settlement in 2015, both related to deceptive pricing. Here, The Children’s Place will distribute up to 800,000 vouchers worth a total of $4.8 million to class members. Depending on the number of claimants who avail themselves of the vouchers, the retailers may also offer vouchers worth 25% off a purchase of up to $100, which could bring up the settlement value to $20 million. Additionally, it has agreed to pay attorneys’ fees for class counsel of about $1 million, and up to an additional $1 million for administrative fees.
Dismissed/Stayed
Federal Judge Dismisses Dannon “Natural” Yogurt Labeling Suit
On December 4, 2017, a New York federal judge granted Dannon’s motion to dismiss a false labeling product liability class action suit against them. The class action suit alleged that Dannon yogurt is falsely labeled as “natural” since the cows may have eaten genetically modified feed, or have been subjected to non-natural processes to increase milk yield, such as being given antibiotics. The suit alleged violations of Minnesota’s deceptive trade practices act and false advertising law, New York’s common law fraud claim, and breach of warranty under the laws of more than 40 states. In her opinion, U.S. District Judge Katherine Forrest held that there was no legal support for the idea that a cow that eats GMO feed, or is subjected to various animal husbandry practices, produces “unnatural” milk. Additionally, the court found that Dannon did and does not represent that its products are GMO-free or free from antibiotics or hormones. Podpeskar v. Dannon Company Inc., Case No. 1:16-cv-08478, U.S. District Court for the Southern District of New York.
FTC and NY AG’s False Advertising Claims Against Prevagen Dismissed
On September 28, 2017, a New York federal judge granted Prevagen’s motion to dismiss fraud and false advertising claims brought against them by the Federal Trade Commission and New York Attorney General Eric Schneiderman. The New York AG’s office brought false advertising and fraudulent acts claims while federal and state regulators accused the makers of Prevagen of violating FTC regulations over false proof and unsubstantiated efficacy claims. U.S. District Judge Louis Stanton found that regulators’ claims that a study showing Prevagen’s primary ingredient, apoaequorin, was clinically proven to help improve memory showed no statistically significant improvement was untrue. Stanton dismissed the FTC allegations for failure to state a claim. Because the federal claims were dismissed, Stanton declined to exercise jurisdiction, and has left it to state courts to decide whether the case has merit, should regulators seek action there. Federal Trade Commission v. Quincy Bioscience Holding Co., Case No. 17-cv-00124, U.S. District Court for the Southern District of New York.
For more information, contact: Kate Watkins
REGULATORY ROUNDUP
With a new executive administration getting its bearings in Washington, the regulatory landscape is in a state of flux—with important changes on the horizon. Each newsletter we bring you a rundown of key developments for the consumer products industry from each of the three main regulatory agencies, as well as the NAD.
CPSC
- On October 26, 2017, CPSC issued a Final Decision and Order holding that Zen Magnets and Neoballs rare-earth magnet sets (Zen Magnets) are a substantial product hazard. CPSC ordered a stop sale of Zen Magnets and ordered the parties to submit to the Commission within 30 days a proposed corrective action plan that includes a refund and notice to the public. The Commission’s Final Decision and Order sets aside a March 2016 administrative law judge’s ruling that Zen Magnets did not present a substantial product hazard when accompanied by warnings and proper age recommendations and that had ordered only a partial recall of Zen Magnets.
- The U.S. Consumer Product Safety Commission (CPSC) voted 3 to 2 on October 18, 2017, to issue a final rule prohibiting children’s toys and child care articles containing more than 0.1 percent of certain phthalate chemicals. On October 27, 2017, CPSC published a final rule, Prohibition of Children's Toys and Child Care Articles Containing Specified Phthalates, the prohibition affects children's toys and child care articles that contain concentrations of more than 0.1 percent of diisononyl phthalate (DINP), diisobutyl phthalate (DIBP), di-n-pentyl phthalate (DPENP), di-n-hexyl phthalate (DHEXP), or dicyclohexyl phthalate (DCHP). This rule fulfills CPSC’s requirement to promulgate a final rule after receiving the final Chronic Hazard Advisory Panel (CHAP) study on the effects on children's health of all phthalates and phthalate alternatives as used in children's toys and child care articles and CHAP’s report providing recommendations to the CPSC regarding whether any phthalates or phthalate alternatives, other than those already permanently prohibited, should be prohibited. The rule will take effect 180 days after publication, on April 25, 2018.
- On October 27, 2017, CPSC issued a notice of proposed rulemaking that would update the existing notice of requirements (NOR) for prohibitions of children's toys and child care articles containing specified phthalates that provide the criteria and process for Commission acceptance of accreditation pursuant to the Consumer Product Safety Act (CPSA). The proposed NOR would revise the current NOR to be consistent with the final phthalates rule, described above and codified in the Code of Federal Regulations (CFR). Comments are due by January 10, 2018.
- On November 17, 2017, CPSC released its Agency Financial Report for Fiscal Year 2017. The purpose of the U.S. Consumer Product Safety Commission’s Fiscal Year (FY) 2017 Agency Financial Report (AFR) is to assist Congress, the President, and the American people in assessing the agency’s stewardship of the resources it is provided. This annual report is required by legislation and complies with the requirements of the Office of Management and Budget’s Circulars A-11, Preparation, Submission, and Execution of the Budget, and A-136, Financial Reporting Requirements. An Annual Performance Report (APR), which focuses on strategic goals and performance results will be published in February 2018. The FY 2017 APR will provide more detailed performance information and analysis of performance results.
FDA
- On Wednesday, October 25, 2017, Dr. Scott Gottlieb, commissioner of the Food and Drug Administration, proposed his agency take a more active role combating opioid drug abuse in the U.S., including urging greater use of addiction-treatment medicines. He told members of the House Committee on Energy and Commerce at a Wednesday hearing on narcotic painkillers that such a role for the FDA, which could include convening meetings to discuss the evidence of treatment benefits from drugs like naltrexone and buprenorphine, is a response to the seriousness of the current opioid-addiction crisis. The following day, President Donald Trump declared opioid addiction a “public health emergency” as he sought to accelerate a federal government response to the crisis. President Trump also said his administration was looking at bringing lawsuits against unspecified “bad actor” companies. President Trump’s declaration did not include a commitment to new funding.
FTC
- On October 19, 2017, the White House announced that President Trump picked Joseph J. Simons, a veteran antitrust lawyer who has represented tech giants like Microsoft, to lead the Federal Trade Commission. Mr. Trump has also chosen Noah Phillips, chief counsel for Senator John Cornyn, Republican of Texas, and Rohit Chopra, a fellow at a consumer advocacy group, to fill the remaining two seats at the agency. The timing of the official nominations is unclear. They will be reviewed by Congress but are expected to be approved.
- On November 30, 2017, Health Research Laboratories, LLC and its owner Kramer Duhon (collectively, HRL) agreed to settle several deceptive advertising charges by the FTC and the State of Maine. HRL marketed two products, BioTherapex and NeuroPlus with unsubstantiated health claims, and also offered customers “risk-free trial offers” that came with undisclosed requirements attached, including having the customer pay shipping costs, and a deceptively short trial period (e.g., the trial was for 30 or 60 days, but the 10–14 days the product took to arrive ate into that trial period). The settlement bans the defendants from making any of the FTC’s seven “Gut Check” weight loss claims, requires human clinical testing to support future health-related claims, requires competent and reliable scientific evidence to support other health benefit claims, prohibits defendants from misrepresenting the terms of free or risk-free trial offers, requires defendants to get consumers’ consent for negative option offers prior to using consumers’ billing information to obtain payment, and prohibits misrepresentations about consumer or expert endorsers. The order imposes a judgment of $3.7 million, which will be suspended upon payment of $800,000.
- On December 4, 2017, the FTC announced that it will hold a public workshop in Washington, DC, on March 7, 2018 “to explore issues regarding competition in the contact lens marketplace, consumer access to contact lenses, prescription release and portability, and related subjects.” This workshop comes during the Commission’s regulatory review of the Contact Lens Rule. The workshop will cover topics including: consumers’ ability to comparison shop for contact lenses; the use of electronic health records, patient portals, and other technology to improve prescription portability; the interaction between the Contact Lens Rule and emerging telehealth business models; the potential for new technology to improve the prescription verification process; and modifications to the Rule to foster competition and maximize consumer benefits, including benefits to eye health.
- On December 12, 2017, the FTC hosted an Informational Injury workshop discussing consumer injury in the context of privacy and data security. Panelists discussed how to characterize these injuries, how to measure them, and what factors businesses and consumers consider when evaluating the tradeoffs between providing consumer information and potentially increasing their exposure to injuries.
NAD
- On November 14, 2017, the Beech-Nut Nutrition Company has said it will discontinue all advertising claims challenged by Nestle Nutrition U.S. before the National Advertising Division. Nestle, maker of Gerber infant foods, challenged claims made in advertising for Beech-Nut infant cereal products. In response to NAD’s initial inquiry, Beech-Nut said that it had decided for unrelated business reasons to permanently discontinue several of the claims at issue before Gerber filed a challenge. Further, Beech-Nut stated that it has discontinued all of the advertising at the center of the challenger’s complaint and argued that NAD should administratively close the proceedings. NAD determined that many of the claims continued to appear in the marketplace after it opened its inquiry. NAD declined to administratively close the matter. However, in reliance on the advertiser’s representation that the claims have been permanently discontinued, NAD did not review the claims on their merits. The voluntarily discontinued claims will be treated, for compliance purposes, as though NAD recommended their discontinuance and the advertiser agreed to comply.
- The National Advertising Division has recommended that Too Faced Cosmetics, LLC, discontinue the “1,944% more volume” claim – and before and after photographs – made on product packaging and online videos for the company’s Better Than Sex original and waterproof mascara. NAD reviewed Too Faced Cosmetics’ testing and found that the testing could not support performance claims because the methodology was not consumer relevant. NAD also determined that the advertiser’s “before” and “after” images reasonably conveyed a message that consumers using the product will achieve similar eyelash volume when they apply the product according to its use instructions without reliable evidence in the record demonstrating that consumers will achieve a similar eyelash volume – a claim not supported by Too Faced’s testing. The company said it will appeal NAD’s decision.
For more information, contact: Senior Law Clerk Jessica Gilbert
Crowell & Moring’s Retail & Consumer Products Law Observer
Each week, Crowell & Moring’s Advertising & Product Risk Management Group brings you the top stories in retail and consumer products law.
ROSCA Enforcement Ahead: FTC Settles with AdoreMe for $1.38 Million
With the explosion of subscription business models, consumer complaints have skyrocketed as well. Lauren Aronson discusses the recent FTC settlement with subscription lingerie service AdoreMe.
“Do Not Resuscitate”: Lessons for Advertisers
Can advertisers be taken at their word? Chris Cole draws a parallel between “Do Not Resuscitate” tattoos and the implication of advertising claims.
Soda Stays Safe in San Francisco
Michelle Gillette and Kimberley Johnson provide an update on the war on sugar, discussing the Ninth Circuit’s blockage of the sweet drink warning labels mandate.
Court Dismisses FTC’s Unfairness Claims Against D-Link
D-Link is a manufacturer of routers and IP cameras. Danielle Rowan breaks down the Northern District of California’s dismissal of FTC’s dismissal of unfairness claims against D-Link and allowance of deception claims.
Chris Cole explains what the FTC and NYAG suit against Quincy Biosciences, the maker of Prevagen, means for advertising of dietary supplements and related health claims.
It’s Only A Game. Or Is It? Advergaming In The Digital Age
Consumers are winning in the digital age. And marketing teams are being forced to think outside of the box. Chalana Williams discusses the legal risks of advertising through mobile gaming apps.
Report on the Autonomous Vehicle Safety Regulation World Congress 2017
Cheryl Falvey provides a report on key themes and takeaways from the 2017 Autonomous Vehicle Safety Regulation World Congress.
Subverting Democracy, Advertising, and the Economy Through Bots
Chris Cole explains the use of non-human bots for ad fraud and how bots can generate what appear to be valid impressions, thereby triggering payment to advertisers.
CROWELL & MORING SPEAKS
Upcoming Engagements
ACI Advertising Claims Substantiation Boot Camp, January 24-26, 2018 – New York, NY
- Chris Cole will be speaking at this inaugural event as part of a panel entitled “Put it to the Test: Evaluating the Degree of Substantiation Necessary to Back Your Claim.” Click here for more information and to register for the conference.
2018 FBA Fashion Law Conference, February 9, 2018 – New York
- Frances Hadfield will be speaking on a panel titled “Trump’s NAFTA Renegotiation: Potential Changes and Impacts to Brands, Apparel and Textiles.” Click here for more information and to register for the conference.
2018 ICPHSO Annual Meeting and Training Symposium, February 21, 2018 – Orlando
- Matthew Cohen will be moderating the plenary session titled “ICPHSO and Product Safety: A Travel Through Time” discussing the dramatically changed product safety landscape in honor of ICPHSO’s 25th anniversary. Click here for more information and to register for the conference.
ABA Section of Antitrust Law 2018 Spring Meeting, April 11-13, 2018 – Washington, DC
- Chris Cole will be speaking at the annual Spring Meeting as part of a panel discussing practical strategies for defending multi-jurisdictional investigations enforcement, and private litigation.
Previous Engagements
Cyber Reputation Defense, October 10, 2017 – Webinar
- On October 10, Cliff Zatz, Laura Aradi, Joe Meadows, and Chalana Williams presented a webinar on the rise of internet defamation cases and defense against reputational attacks in the cyber-world. Click here to listen to the webinar and for more information.
2017 ANA/BAA Marketing Law Conference, November 13-15, 2017 – Chicago, IL
- Crowell & Moring sponsored the annual ANA/BAA Marketing Law Conference. Chris Cole moderated and Ryan Tisch spoke a panel entitled “The Private Label Revolution.” David Ervin moderated a panel called “The Shared Economy: Disruption Continues.” To learn more about the conference and access presentation materials, click here.
What's Next -- Hot Issues at the Nexus of Consumer Protection, Antitrust, and Sports, December 5, 2017 – Webinar
- Lauren Aronson participated in a webinar on the impact of antitrust and consumer protection in the sports industry and a preview of what is to come in 2018.
FDLI Enforcement, Litigation, and Compliance Conference, December 6-7, 2017 – Washington, DC
- John Fuson spoke on a panel entitled “Management Oversight and Control: How to Ensure Compliance and Limit Liability” at the FDLI enforcement conference focused on the drug, device, food, and tobacco industries.
Intellectual Property and Product Liability Lifecycle Seminar, December 11, 2017 – Israel
Crowell & Moring and ISUS IP are embarking on a new partnership to provide unique offerings to clients. Andrew Kaplan spoke at the inaugural event, a case study seminar examining a product lifecycle from conception through launch of a hypothetical Israeli product and company entering the US market.
1 The Federal Aviation Administration (FAA) recently announced that it also is working on a comprehensive scheme to regulate 3D printing materials and processes in the aerospace sector.
2 See Technical Considerations for Additive Manufactured Medical Devices: Guidance for Industry and Food and Drug Administration Staff (officially dated Dec. 5, 2017), available at https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM499809.pdf.
3 See Statement by FDA Commissioner Scott Gottlieb, M.D., on FDA ushering in new era of 3D printing of medical products; provides guidance to manufacturers of medical devices (Dec. 4, 2017), available at https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587547.htm.
4 https://ntl.bts.gov/lib/52000/52800/52889/812075_CybersecurityBestPractices.pdf.
5 https://www.federalregister.gov/documents/2017/10/27/2017-23267/prohibition-of-childrens-toys-and-child-care-articles-containing-specified-phthalates.
6 See e.g., In re: Whole Foods Mkt., Inc., Greek Yogurt Mktg. and Sales Practices Litig., Case No. 1:14-mc-02588-SS (W.D. Tex. Feb. 16, 2016), ECF No. 47, available here (rejecting plaintiffs’ argument that their claims were not preempted because, while the protocol outlined in 21 C.F.R. § 101.9(g)(2) mandates the testing methodology to be used by the FDA, the regulations do not require plaintiffs in a private lawsuit to follow that methodology).
Contacts






Insights

Client Alert | 1 min read | 04.18.24
GSA Clarifies Permissibility of Upfront Payments for Software-as-a-Service Offerings
On March 15, 2024, the General Services Administration (GSA) issued Acquisition Letter MV-2024-01 providing guidance to GSA contracting officers on the use of upfront payments for acquisitions of cloud-based Software-as-a-Service (SaaS). Specifically, this acquisition letter clarifies that despite statutory prohibitions against the use of “advance” payments outside of narrowly-prescribed circumstances, upfront payments for SaaS licenses do not constitute an “advance” payment subject to these restrictions when made under the following conditions:
Client Alert | 4 min read | 04.18.24
Client Alert | 6 min read | 04.16.24
Navigating the AI Intellectual Property Maze - Key Points From Congressional Hearing
Client Alert | 5 min read | 04.15.24
Making the EU Courts More Efficient for Trade-Related Decisions